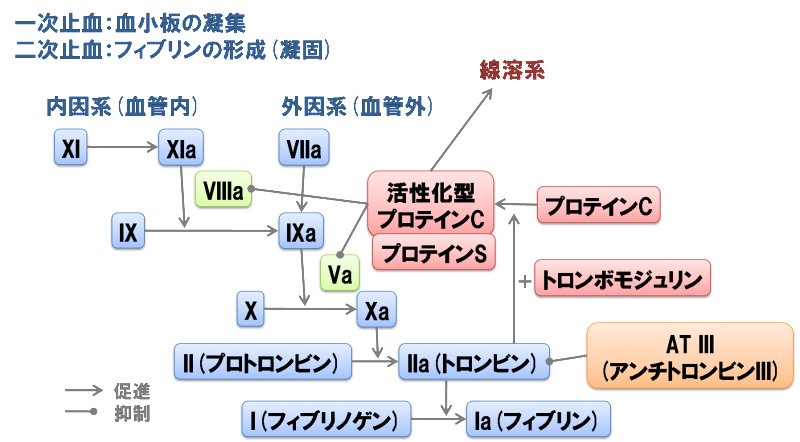
プロテインC(英: protein C、別名: 第XIV因子(blood coagulation factor XIV)、autoprothrombin IIA)は酵素前駆体で、タンパク質分解を経て活性化されたプロテインC(活性化プロテインC、APC)はヒトや他の動物において抗血液凝固作用、炎症、細胞死、血管の透過性の維持に重要な役割を果たす。APCは、主に活性化された第V因子と第VIII因子(それぞれ第Va因子と第VIIIa因子と呼ばれる)をタンパク質分解によって不活性化することで、これらの機能を果たす。APCはその活性部位にセリン残基を含むため、セリンプロテアーゼに分類される。プロテインCは、ヒトでは2番染色体のPROC遺伝子にコードされる。
酵素前駆体型のプロテインCは血漿中を循環するビタミンK依存性糖タンパク質であり、ジスルフィド結合で連結された軽鎖と重鎖の2本のポリペプチド鎖から構成される。プロテインC酵素前駆体は、血液凝固に深く関わるタンパク質トロンビンに結合することで活性化される。活性化はトロンボモジュリンと血管内皮細胞プロテインC受容体(EPCR)の存在によって大きく促進される。EPCRは血管内皮細胞の細胞表面に発現しているため、APCは主に血管内皮細胞の近傍に存在し、血管内皮細胞と白血球に影響を与える。プロテインCの重要な役割は抗血液凝固作用であり、プロテインCの欠乏あるいはAPCに対する抵抗性によって、危険な血栓の形成(血栓症)のリスクが増大する。
組換え型ヒト活性化プロテインC製剤はドロトレコギンアルファ (活性型)という名称で知られ、イーライリリー・アンド・カンパニーによってザイグリス(Xigris)の商標名で開発・販売された。しかし、その臨床使用へ向けた研究には大きな論争が伴っている。イーライリリーは重症敗血症や敗血症性ショックの患者に対する使用を促進する積極的な広告宣伝活動を行い、2004年のSurviving Sepsis Campaignのガイドラインを後援した。しかし2012年のコクランレビューでは、生存率は改善されず出血リスクを増大させるため使用は推奨されないとされた。2011年10月、成人を対象とした治験で高い死亡率がみられたためザイグリスは市場から撤退した。
歴史
ヒトの体内でのプロテインCの抗血液凝固活性はSeegersらによって初めて記載され、そこではプロテインCにはautoprothrombin II-aという名称が付けられていた。プロテインCは1976年にJohan Stenfloによってウシの血漿から初めて単離され、ビタミンK依存性タンパク質であることが明らかにされた。彼は、DEAE-Sepharoseを用いたイオン交換クロマトグラフィーにおいて3番目に溶出するピーク("ピークC")に含まれるタンパク質であったため、プロテインCと名付けた。当時Seegersは凝血機能を測定する一般的なアッセイでは検出されないビタミンK依存性凝固因子を探索しており、Stenfloが発見したタンパク質が自身の記載したものと同一であることを発見した。続いてAPCが発見され、1977年にはAPCが第Va因子を不活性化することが初めて認識された。1980年にVeharとDavieはAPCが第VIIIa因子の不活性化も行うことを発見し、その直後にWalkerによってプロテインSがAPCのコファクター(補因子)であることが示された。1982年Griffinらによる家系分析によって、プロテインC欠乏症と静脈血栓塞栓症の症状との関係性が初めて示された。ホモ接合型のプロテインC欠乏症とその健康への重篤な影響がいくつかのグループによって1984年に記載された。プロテインCのcDNAクローニングは1984年にBeckmannらによって行われ、肝臓でのプロテインCの産生を担う遺伝子領域のマッピングが行われた。1987年のTaylorらによる一連の研究によって、APCは血液凝固障害を防ぎ、致死量の大腸菌E. coliを注入されたヒヒの死を防ぐ効果があることが示された。
1993年、Dahlbäckらによって遺伝的要因によるAPC抵抗性が発見され、家族性の血栓性素因との関係が示された。1994年に、APC抵抗性をもたらす変異としては一般的な第V因子ライデン変異がBertinaらによって記載された。2年後、Glaドメインを除くAPCの構造が2.8 Åの分解能で明らかにされた。2001年にはPROWESS(Recombinant Human Activated Protein C Worldwide Evaluation in Severe Sepsis)試験が開始され、APCの注入によって敗血症の症状の多くが緩和され、敗血症患者の致死率が大きく低下することが示された。その年の末には、組換えヒトAPC製剤ドロトレコギンアルファ(活性型)がアメリカ食品医薬品局(FDA)によって重症敗血症の治療に承認された最初の薬剤となった。しかし、2011年に成人を対象とした治験で高い死亡率がみられたため、市場から撤退した。2002年、プロテインCがプロテアーゼ活性化受容体1(PAR-1)を活性化することを示す論文がサイエンス誌に発表され、このタンパク質による免疫系の調節機構である可能性がある。
遺伝子
プロテインCは、ヒトでは"protein C (inactivator of coagulation factors Va and VIIIa)"と命名された遺伝子にコードされている。HUGO遺伝子命名法委員会によって承認された遺伝子シンボルは"PROC"(protein Cに由来)である。PROC遺伝子は2番染色体(2q13-q14)に位置し、9つのエクソンから構成され、そのヌクレオチド配列の長さは約11,000塩基である。
構造
ヒトのプロテインCはビタミンK依存性糖タンパク質である。構造的には、プロトロンビン、第VII因子、第IX因子、第X因子といった血液凝固に影響を与える他のビタミンK依存性タンパク質と類似している。プロテインCの合成は肝臓で行われ、32アミノ酸からなるN末端のシグナルペプチドを含む1本鎖のタンパク質(プレプロプロテインC、preproprotein C)として合成される。プロテインCはLys198とArg199の2ペプチドの除去によって形成され、N-結合型炭水化物で修飾された重鎖(41 kDa)と軽鎖(21 kDa)からなるヘテロ二量体へと変化する。重鎖と軽鎖はCys183とCys319の間のジスルフィド結合によって連結されている。
不活性な前駆体型プロテインCは419アミノ酸からなり、複数のドメインで構成される。Glaドメイン(43–88番残基)、ヘリカル芳香族セグメント(89–96)、2つのEGF様ドメイン(97–132、136–176)、活性化ペプチド(200–211)、トリプシン様セリンプロテアーゼドメイン(212–450)で構成される。軽鎖はGlaドメイン、EGF様ドメイン、芳香族セグメントを含む。重鎖はプロテアーゼドメインと活性化ペプチドを含む。プロテインCの85–90%はこの形の酵素前駆体として血漿中を循環し、活性化に備えている。残りのプロテインC前駆体はわずかに変化した形態で存在する。プロテインCの活性化は、トロンビンによる重鎖のN末端から活性化ペプチドの切除によって行われる。プロテインCの活性部位はセリンプロテアーゼに典型的な触媒三残基(His253、Asp299、Ser402)を含む。
生理学
プロテインCの活性化は、トロンボモジュリンとEPCRによって強く促進される。EPCRは主に血管内皮細胞(血管の内側を構成する細胞)に存在している。トロンボモジュリンの存在は活性化を数桁のオーダーで加速させ、EPCRは活性化を20倍加速する。これらの2つの因子のいずれも存在しない場合、マウスでは過剰な血液凝固のため胎生致死となる。内皮細胞では、APCは血液凝固、炎症、細胞死(アポトーシス)の調節に主要な役割を果たす。トロンボモジュリンによる強力な活性化促進効果のため、プロテインCはトロンビンではなく、トロンビン-トロンボモジュリン(さらにはトロンビン-トロンボモジュリン-EPCR)によって活性化されると言われることもある。活性型となったAPCはEPCRに結合したままである場合もない場合もあり、EPCRとの親和性は前駆体と同程度である。
前駆体型のプロテインCは、正常な成人の血漿中に65–135 IU/dLの濃度範囲で存在する。APCのレベルはこれよりも約2000倍低い。軽度のプロテインC欠乏症は20 IU/dL以上通常以下、中等度の欠乏症は1–20 IU/dL、重症欠乏症は1 IU/dL以下または不検出に相当する。プロテインCの濃度は生後6ヶ月まで上昇を続け、平均60 IU/dLのレベルに達する。その後の小児期は低いままであり、思春期の後に成人のレベルに到達する。APCの半減期は約15分である。
経路
APCの発現レベルと体内での活性は、特定の化学反応によって制御されている。プロテインCは多機能タンパク質で、抗血液凝固作用と細胞保護作用(細胞に対する作用)という2つの主要な機能が存在する。プロテインCがどちらの機能を発揮するかはAPCが活性化後もEPCRに結合したままであるかどうかに依存しており、抗血液凝固作用はAPCがEPCRに結合していないときに生じる。この場合、プロテインCは第Va因子と第VIIIa因子をタンパク質分解によって不可逆的に不活性化する抗血液凝固因子として機能し、それぞれ不活性な第Vi因子と第VIIIi因子に変化させる。プロテインCがEPCRに結合したままの場合は、APCはエフェクター基質であるPAR-1に作用することで細胞保護効果を発揮する。APCの抗血液凝固作用と細胞保護作用はある程度独立した現象であり、一方の経路の発現が他方の影響を受けることはない。
プロテインCの活性は、トロンボモジュリンとEPCRのいずれかの減少によってダウンレギュレーションされる可能性がある。この調節は、IL-1βやTNF-αなどの炎症性サイトカインによって行われている可能性がある。活性化された白血球は炎症時に炎症メディエーターを放出し、トロンボモジュリンとEPCRの双方の合成を阻害するとともに、内皮細胞表面からのEPCRの切除を誘導する。これらの双方の作用がプロテインCの活性をダウンレギュレーションする。トロンビンもEPCRのレベルに影響を与える。加えて、好酸球などの細胞から放出されるタンパク質もプロテインCの活性化を阻害し、これは特発性好酸球増加症候群による心疾患でみられる血栓症の発症機構である可能性がある。また、プロテインCはプロテインCインヒビターによっても阻害される。プロテインCは血小板第4因子によってアップレギュレーションされる可能性がある。このサイトカインは、プロテインCのGlaドメインとトロンボモジュリンのグリコサミノグリカン(GAG)ドメインとの間で静電的相互作用を形成し、活性化反応のミカエリス定数(KM)を低下させることによってプロテインCの活性化を促進すると推測される。
抗血液凝固作用
プロテインCは人体における抗血液凝固機構の主要な構成要素である。プロテインCはセリンプロテアーゼ前駆体であり、APCとなって第Va因子や第VIIIa因子のペプチド結合の分解を行う。第Va因子と第VIIIa因子は、トロンビンの産生に関して強い凝固促進作用を持つコファクターで、血液凝固における重要な要素である。第Va因子と第VIIIa因子の不活性化に関与するコファクターには、プロテインS、第V因子、高密度リポタンパク質、アニオン性リン脂質とスフィンゴ糖脂質がある。
第Va因子はプロトロンビンと第Xa因子(活性化第X因子)に結合し、トロンビンの産生速度を4桁(1万倍)のオーダーで増加させる。そのため、第Va因子の不活性化によってトロンビンの産生は事実上停止する。一方、第VIIIa因子は第Xa因子の産生のコファクターである。第VIIIa因子は第X因子の活性化を約20万倍増加させる。第VIII因子は抗血友病因子(anti-haemophilic factor)としても知られており、第VIII因子の欠乏は血友病Aの原因となる。
APCは、第Va因子を3ヶ所(Arg306、Arg506、Arg679)で切断することとで不活性化を行う。Arg306とArg506の切断は第Xa因子への結合を低下させる。Arg306での切断反応の進行は遅いものの、第V因子の機能調節には必要である。プロテインSはArg306での切断を触媒することでこの過程を促進し、その結果第V因子のA2ドメインが解離する。プロテインSは第Xa因子にも結合し、第Xa因子による第Va因子不活性化過程の妨害を除去する。
第VIIIa因子の不活性化についてはよく理解されていない。第VIIIa因子の半減期は、第IXa因子(活性化第IX因子)による安定化がない場合、約2分である。APCによる第VIIIa因子の不活性化の意義には一部から疑問が投げかけられており、第V因子やプロテインSがどの程度このタンパク質分解のコファクターとして機能しているかは未知である。APCが第VIIIa因子のArg336とArg562の2ヶ所を切断することが知られており、第VIIIa因子を不活性化して第VIIIi因子へ変換するためには、いずれかの切断で十分である。
細胞保護作用
APCがEPCRに結合している際には多数の重要な細胞保護機能を発揮するが、その大部分はEPCRとPAR-1を必要とすることが知られている。そのようなAPCの機能としては、遺伝子発現の調節、抗炎症効果、抗アポトーシス効果、血管内皮バリア機能の保護がある。
細胞をAPCで処理することにより、炎症やアポトーシスの主要経路が遺伝子発現調節を介して効果的に制御されることが示されている。プロテインCによって約20個の遺伝子がアップレギュレーションされ、20個の遺伝子がダウンレギュレーションされることが知られている。前者は主に抗炎症、抗アポトーシス経路に関与するもので、後者は炎症やアポトーシスを促進する傾向がある。APCが遺伝子発現プロファイルを変化させる機構はあまり解明されていないが、少なくとも部分的には転写因子活性の阻害を伴うものであると考えられている。APCによってアップレギュレーションされる重要なタンパク質にはBcl-2、eNOS、IAPがあり、APCによって大きくダウンレギュレーションされる遺伝子にはp53とBaxがある。
APCは血管内皮細胞と白血球に抗炎症効果を示す。APCは血管内皮細胞からの炎症メディエーターの放出を阻害し、血管の細胞接着分子をダウンレギュレーションする。これによって白血球の接着と組織への浸潤が減少し、下層組織の損傷も低減される。APCは内皮細胞のバリア機能を補助し、走化性を低下させる。APCは、サイトカイン応答を低下させることによって白血球でも炎症メディエーターの放出を阻害するが、敗血症時に見られるように、全身の免疫反応を低下させることによって行われている可能性もある。ラットとヒトの研究の双方において、APCがエンドトキシンによる肺損傷や炎症を低下させることが示されている。
APCの抗アポトーシス効果は広く認識されているが、どのようにアポトーシスが阻害されているかの正確な機構は不明である。APCは神経保護効果を示すことも知られている。抗アポトーシス効果はカスパーゼ3とカスパーゼ8の活性化の低下、Bax/Bcl-2比の改善、p53のダウンレギュレーションによって行われていると考えられている。
APCは血管内皮細胞のバリア機能の十分な保護を行う。内皮細胞バリアの乱れとそれに伴う内皮細胞の透過性の増加は、腫脹、低血圧、炎症、そして敗血症の全ての問題と関係している。APCはPAR-1依存的なスフィンゴシンキナーゼ1の活性化を誘導し、スフィンゴシン-1-リン酸をアップレギュレーションすることで内皮細胞のバリア機能を保護する。
いくつかの研究はAPCのタンパク質分解活性が細胞保護機能に寄与することを指摘しているが、一方でタンパク質分解活性のない変異体がin vitroでもin vivoでも細胞保護効果を示すことも報告されている。
疾患における役割
遺伝的なプロテインC欠乏症のうち、単純なヘテロ接合型変異による軽症型では、成人での静脈血栓塞栓症のリスクが大きく増加する。ホモ接合型または複合ヘテロ接合型変異による欠乏症では、子宮内での電撃性紫斑病、重度の播種性血管内凝固症候群と、静脈血栓塞栓症の同時発症がみられることがあり、非常に重篤で通常は致死となる。マウスでのプロテインC遺伝子の欠失は出生前後に致死となる。プロテインCを持たないマウス胎児の初期発生は正常であるが、重度の出血、血液凝固障害、フィブリンの蓄積と肝臓の壊死が起こる。
プロテイン欠乏症は、症状がみられない人の中でも200人から500人に1人の頻度でみられる。対照的に、欠乏症の大きな症状がみられる頻度は2万人に1人である。人種や民族による差は見られない。
APC抵抗性とは、APCがその機能を発揮できない状態である。この疾患の症状はプロテインC欠乏症と類似している。APC抵抗性が生じる変異としてコーカソイドで最も一般的なものは、第V因子のAPC切断部位の変異である。この変異では第V因子のArg506がグルタミン酸に置換されており、第V因子ライデン変異またはR506Q変異と呼ばれる。この変異によって第V因子の切断部位が失われた結果、APCによる第Va因子と第VIIIa因子の双方の効率的な不活性化が事実上停止する。そのため、血液が過剰に凝固しやすい状態となり、常に血栓症のリスクが増大した状態となる。第V因子ライデン変異をヘテロ接合型で有する場合、静脈血栓塞栓症のリスクは通常人と比較して5–7倍、ホモ接合型の場合は80倍高くなる。この変異は、コーカソイドにおける静脈血栓塞栓症の最も一般的な遺伝的要因でもある。
第V因子ライデン変異ほどではないが、他の遺伝的変異もAPC抵抗性を生じさせる。そのような変異には、第V因子の他の部位の変異や、第V因子を標的とする自己抗体の産生、APCのコファクターの機能不全をもたらすものなどが含まれる。また、いくつかの後天的要因によってAPCの抗凝固作用が低下することもある。血栓形成傾向を有する患者の20%から60%には何らかのAPC抵抗性が生じていることが研究から示唆されている。
ワルファリン壊死は、ワルファリンの投与によって生じる後天的なプロテインC欠乏症である。ワルファリンはビタミンKのアンタゴニストでそれ自身抗凝固作用を有するものの、ワルファリンの投与によって電撃性紫斑病と類似した皮膚障害が生じることがある。この反応に類似したものとして、がんに関連した深部静脈血栓症の治療にワルファリンが利用された際に静脈性四肢壊疽が生じることもある。このような場合ワルファリンの投与は、凝固第II因子、第IX因子、第X因子の抑制が生じる前にプロテインC欠乏症が生じないことを確認しながら低用量で再開される。
APCはマラリア原虫Plasmodium falciparumが感染時に放出するヒストンを切断する。切断によってヒストンの炎症促進効果は消失する。
医療における役割
2001年11月FDAは、致命リスクの高い重症敗血症の成人患者に対する治療薬としてドロトレコギンアルファ(活性型)を承認した。ドロトレコギンアルファ(活性型)は組換え型ヒト活性化プロテインC製剤であり、イーライリリー・アンド・カンパニーからザイグリスの商標名で販売された。
ドロトレコギンアルファ(活性型)は出血の増大と、死亡率の低下がみられないこととが判明し、大きな論争の的となった。2011年10月に、ザイグリスは成人を対象とした治験で高死亡率を示したため、市場から撤退した。
肺損傷の患者において、肺の特定部位でのAPCレベルの低下と予後の悪さとの間に相関があることが示されており、APCは肺損傷の治療法としての研究が行われている。またAPCは、動脈閉塞によって脳のある領域の酸素が奪われ組織の細胞死が引き起こされる、虚血性脳卒中の患者の予後の改善のための利用が考えられている。現在唯一承認された治療法である組織プラスミノーゲン活性化因子(t-PA)とAPCとの併用は、低酸素による細胞死を防ぐとともにt-PAの非常に有害な副作用から脳を保護する有望な治療法となる可能性が研究から示唆されている。1型糖尿病の治療時の膵島移植の予後の改善のためのAPCの臨床利用も提唱されている。
脚注
- ^ α: Glaドメインを除去したプロテインCは、82–83番残基間の選択的タンパク質分解によって、Glaドメイン(47–88番残基)の大部分を含むN末端領域を取り除くことで得られた。N末端領域はタンパク質の結晶化を容易にするために除去された。
- ^ β: 過好酸球増加症では、内皮細胞表面の過剰な好酸球特異的顆粒タンパク質(主要塩基性タンパク質(MBP)、好酸球カチオン性タンパク質(ECP)など)がトロンボモジュリンに結合し、トロンボモジュリン表面と静電的相互作用によるプロテインCの活性化への参加を阻害する。
出典
外部リンク
- ペプチダーゼとその阻害因子に関するMEROPSオンラインデータベース: S01.218